Software for numerical simulation of crystal growth by Monte Carlo method.
| Screenshots: |
|
Summary:
- Software name: LeoMonteCrystal.
- Operation system: MS Windows.
- Type: Freeware without limitations.
-
Codes: Complete code project in MS Visual Studio C++ is available to
purchase for learning, modification or implementing of the state of art algorithms
and functions at
LeoMonteCrystal project page.
- Installation: press button to go to
installation page
for obtaining free executable LeoMonteCrystal.
-
Main features:
-
Scientific background:
- A numerical simulation is based on Monte Carlo implementation of modified cellular automaton model. Simulated crystal surface is represented by two dimensional matrix where value and position of the elements define 3D location of the molecule on the surface of the crystal.
- At every next time step each position on the surface of the crystal can accept new molecule and move forward, stay on place or emit molecule and move backward.
- Relative probabilities to accept or emit molecule are calculated on the base of model of thermally activated reaction depending on near surrounding of each molecule on the surface that allows direct introducing of the surface energy into the model of the crystal growth.
- A model also permit calculation of concentration hole type defects formed as result of plugging bottle neck like structures on the rough surface.
-
Setting experimental parameters:
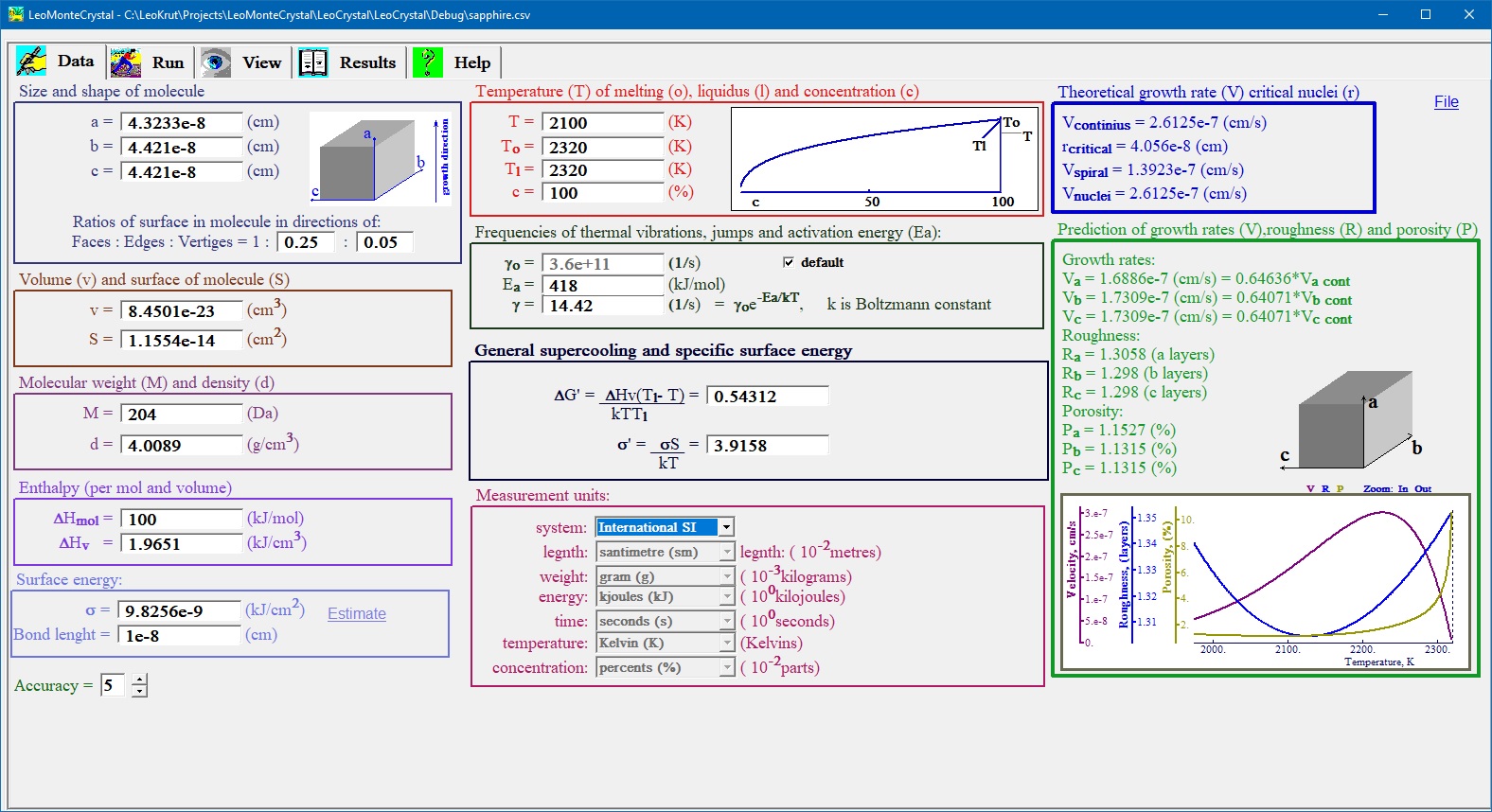
- Parameters of single experiment can be typed in to data panel with flexible choice of to use favorite measurement units.
- LeoMonteCrystal provides unit converter capabilities, permitting to use initial parameters defined in various unit systems as fit a user customary.
- Data input panel instantly displays theoretical values for velocity crystal growth calculated for competitive theoretical models and plus most valuable - with formulas found as result of our research for given set of initial parameters. The temperature dependence of growth rate, surface roughness and defects concentration calculated by our formulas are displayed at separate chart.
- After modification of any one parameters all dependable variables are automatically updated accordingly providing functionality of thermodynamic and crystallography calculator.
- Frequently used set of parameters, corresponded to compound of interest, can be saved or imported to or from coma delimited file (MS Excel compatible).
- There are helpful key shortcuts incorporated in for managing special cases to try for researcher: varying effective size of presumed molecule by multiplying or dividing its dimensions by integer value; estimating value of surface energy; calculating temperature dependence of theoretical values given set of other parameters.
-
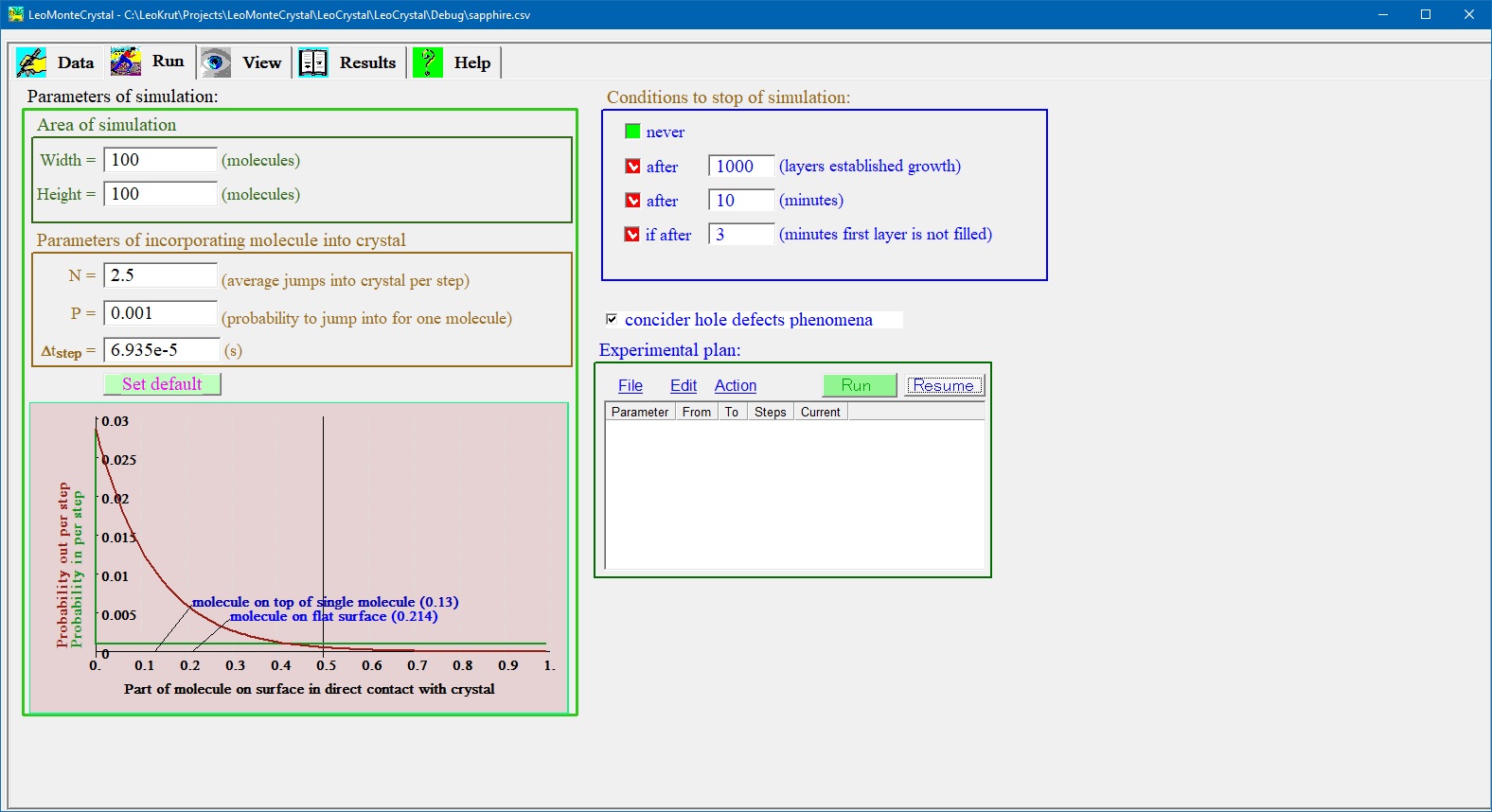
Setting parameters of run for numerical simulation:
- LeoMonteCrystal permits to set size of simulation field and time step for one turn in convenient for user form. One can play from choosing the probability of one molecule to jump into crystalline phase during turn time (or number of such molecules for given size of the field); user also can set conditions to stop a simulation by limiting a time of computer simulation or number of layers filled by molecules.
- On the panel for setting technical parameters for experiment run there is graph presenting probabilities that in time step given position depend on neighbor surrounding on simulation field of crystal surface a molecule will incorporated into crystalline phase or emitted outside or stay unmoved.
- User can run single experiment for thermodynamic and kinetic variables set on Data panel as well to program experimental plan, series experiments flexible varying temperature, surface energy and other parameters.
- Experiment plan can be saved in or open from a file.
-
Visual presentation:
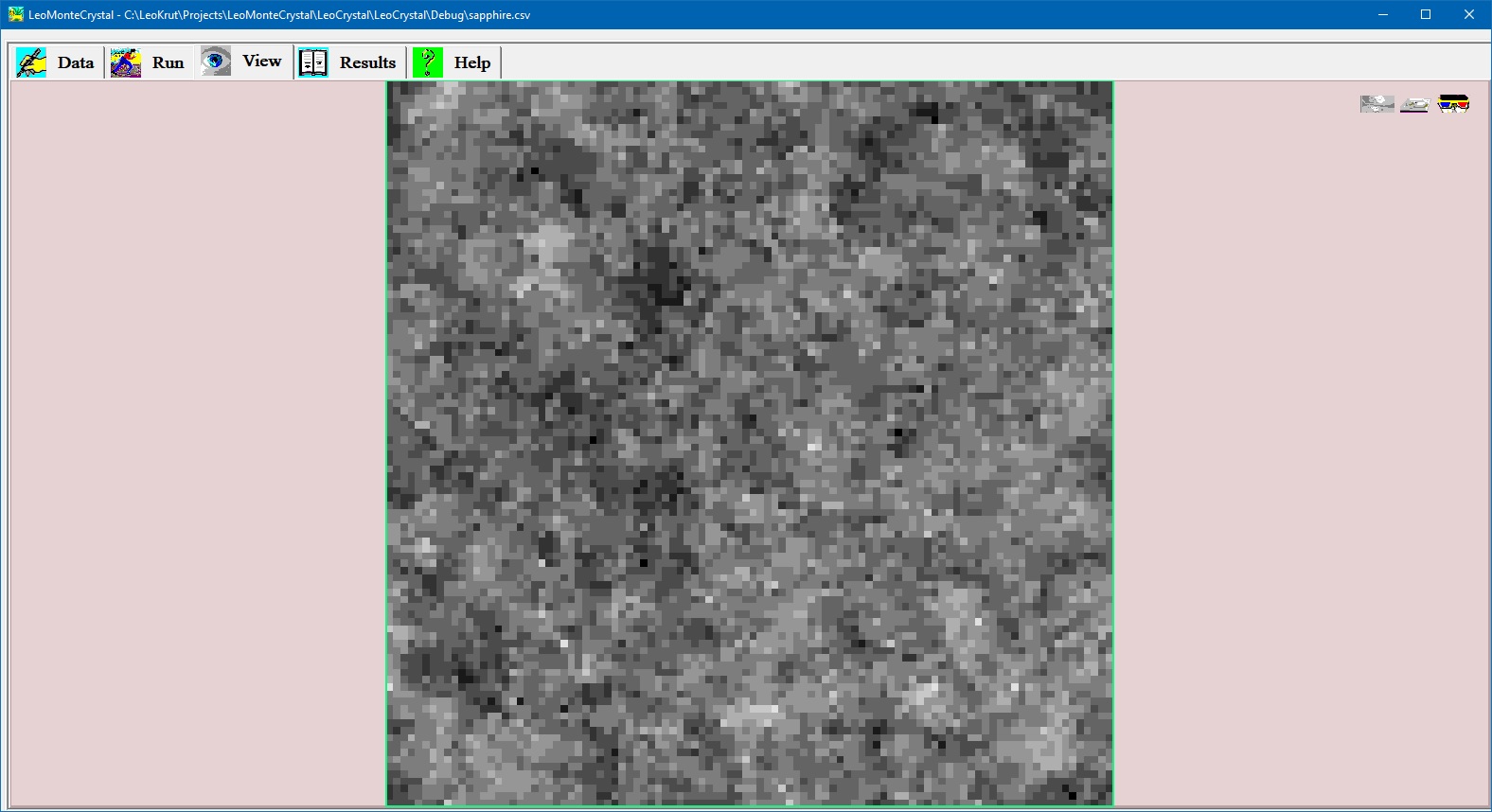
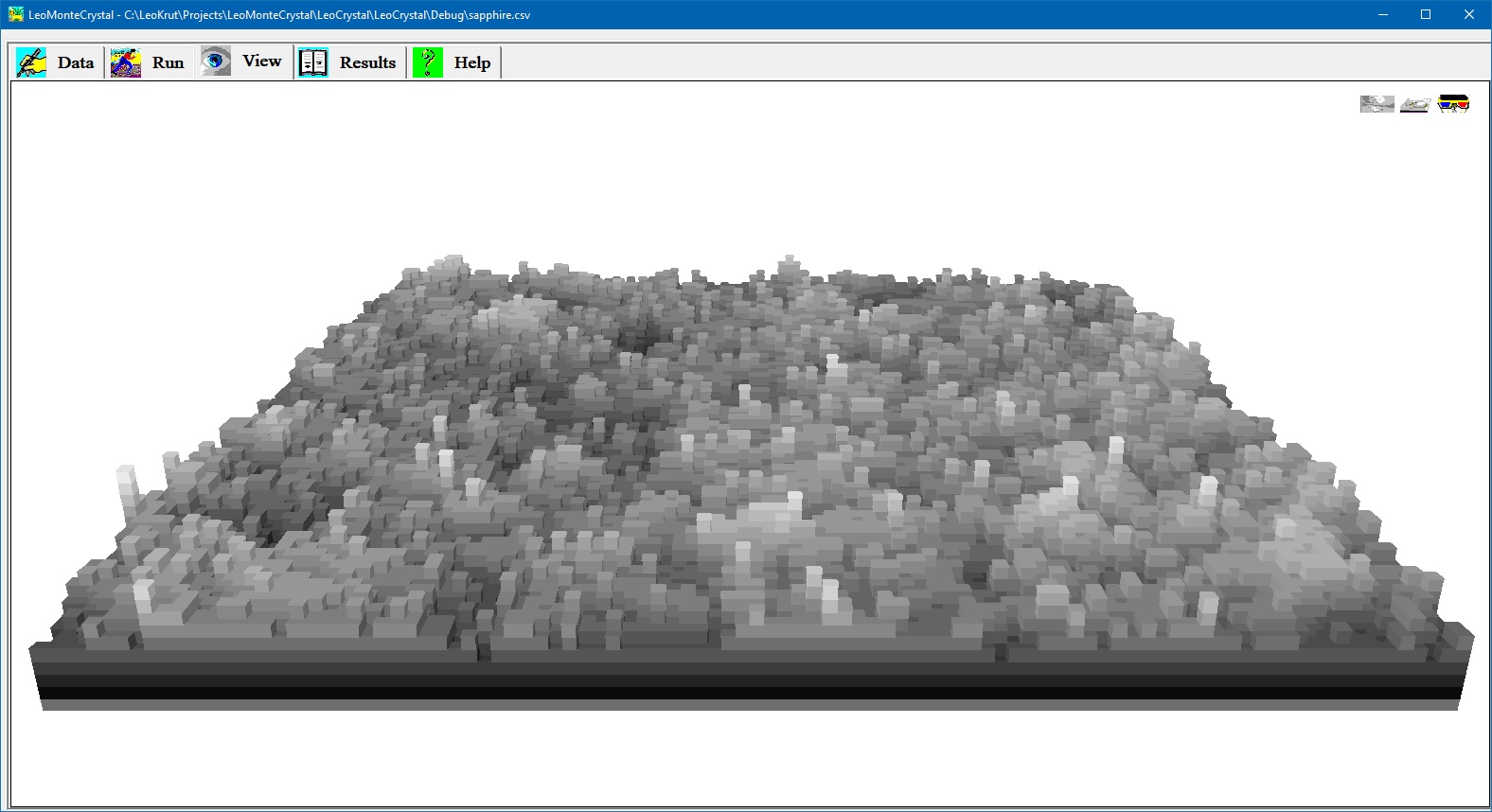
- View panel offers three options to observe surface of the growing crystal during numerical simulation: grey iso-altitude map; bird like view in perspective projection and stereo pair red and blue images to be observed via glasses with corresponding colors.
- User can to move by arrow keys on keyboard position of virtual observer above surface selecting best vantage point.
-
Result charts:
- At the "Results" panel during simulation there are charts displaying timeline of: distance of crystal surface growth progress, its roughness, instant growth rate and concentration of the hole like defects.
- Current characteristics of the numerical simulation: number of time steps, rate of crystal growth, roughness of surface, induction period of critical two dimensional nuclei on smooth surface, concentration of holes and some derivatives from these are displayed in numerical form.
- Depended on simulation conditions type of current mechanism of growth is defined.
- Upon end of experiment conditions of simulation and results of experiment automatically recorded into log file.
-
Help:
- We provide white pages with description of algorithm for numerical simulation, results of our numerical experiments and discovered universal formulas.
- To protect our intellectual property we reserve the right distinct between level of specificity in available for different users supporting papers depend on their verified identity and support of our research.
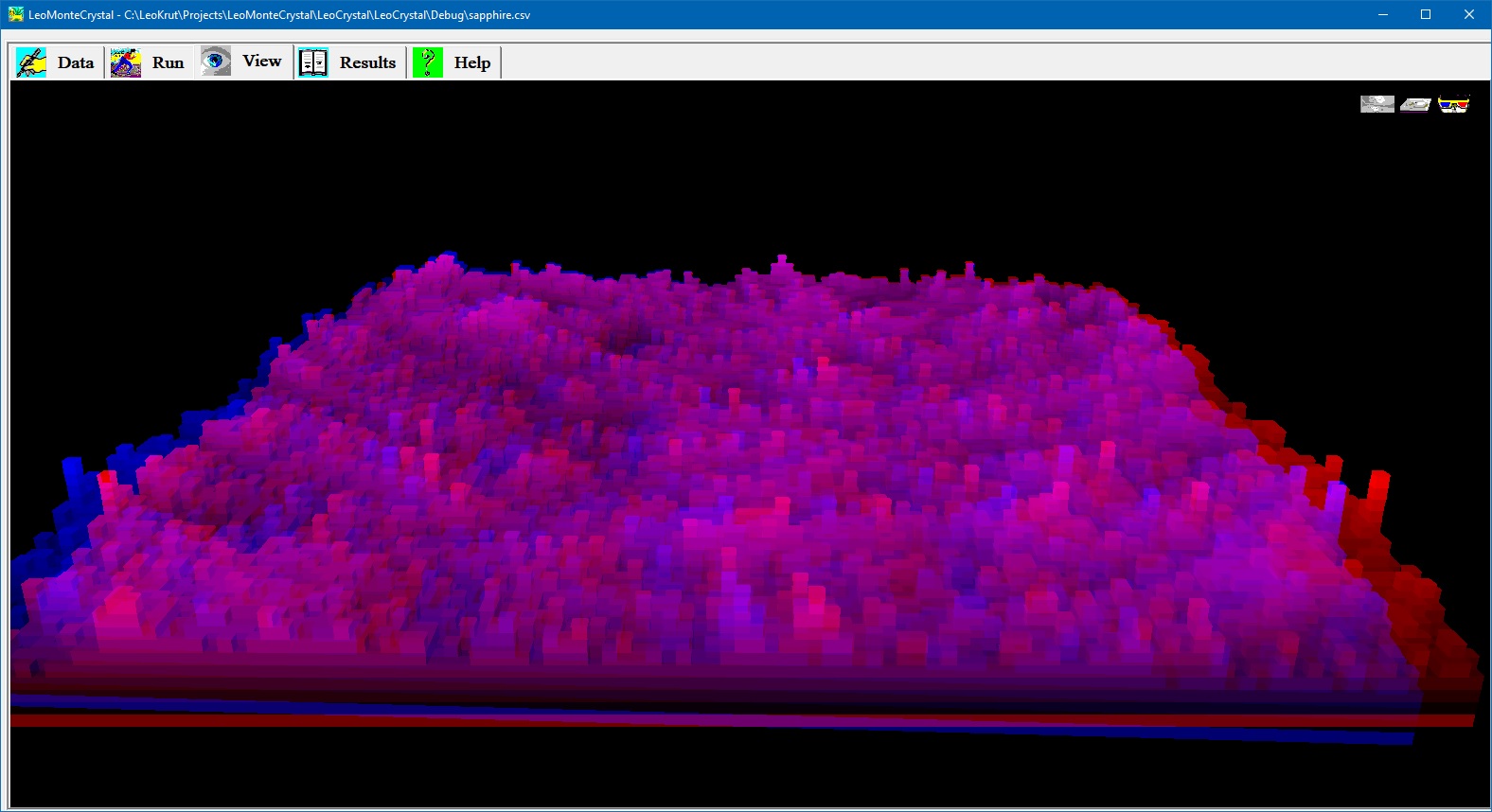
Virtual image of growing crystal surface during numerical simulation. 
View panel permits to observe in real time growing crystal surface during numerical simulation. There are options to see crystal surface as grey iso-altitude map; bird like view in perspective projection and stereo pair red and blue images to be observed via glasses with corresponding colors. Setting parameters and run an experiment in software for numerical simulation of the crystal growth. 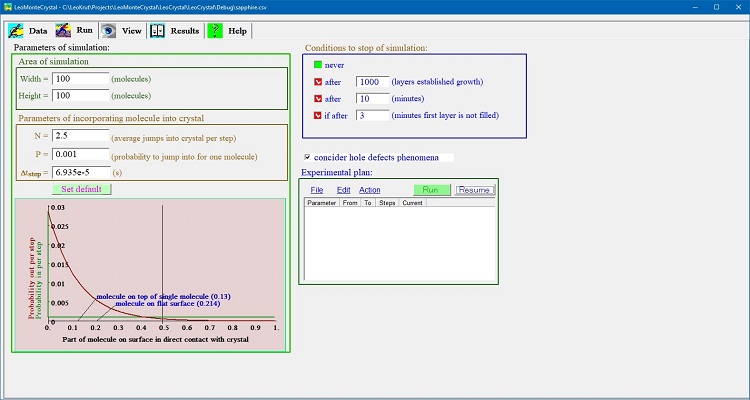
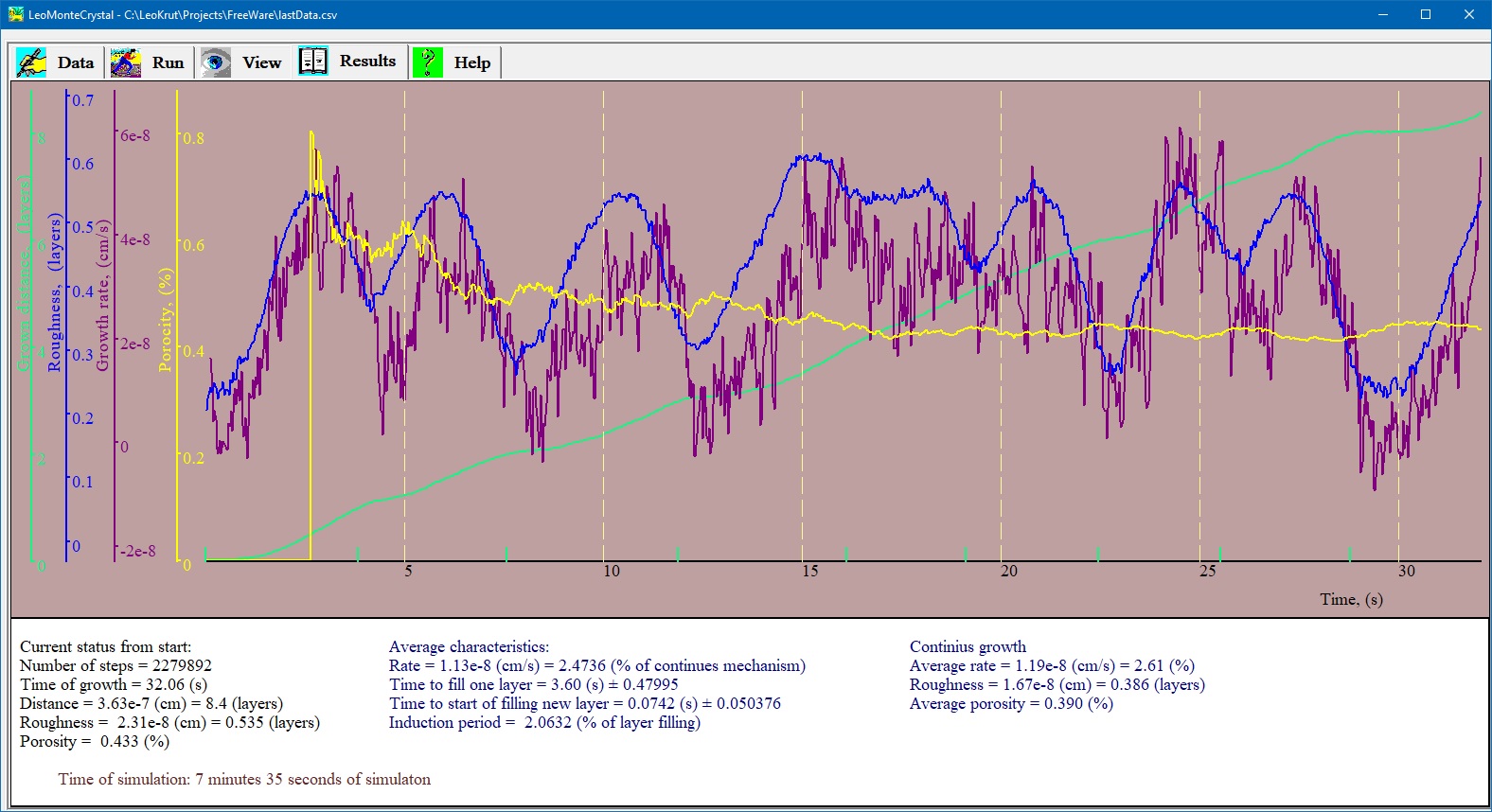
Run panel is for setting parameters of experiment and run it in software for numerical simulation of the crystal growth. User can choose dimensions of simulation field, setting time of one step and design a series of experiment to run automatically varying specific parameters of the process. Presentation of results of numerical simulation of crystal growth. 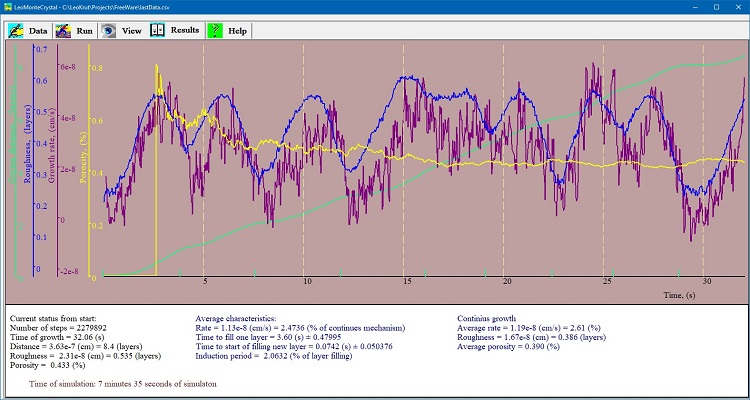
Results panel of LeoMonteCryslal presents of results of numerical simulation of crystal growth in chart form showing advance of surface, growth rate, roughness of surface and concentration of defects in real time during simulation. An interface for data enter in software of numerical simulation of crystal growth. 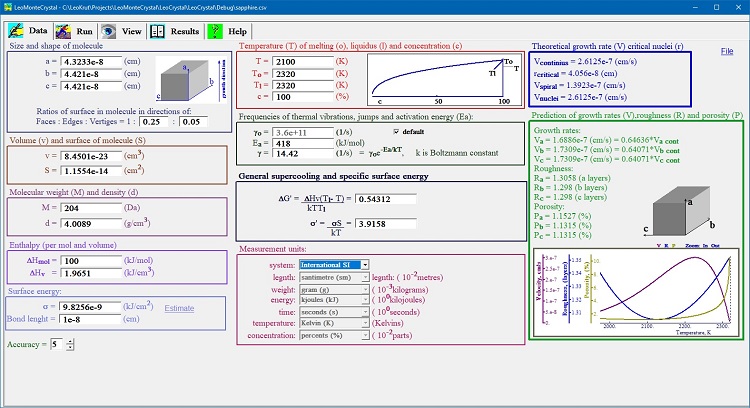
Data entering interface of software of numerical simulation of crystal growth is used for setting thermodynamic and crystallographic parameters of the process.
-
Scientific background: