An interface for data enter in software of numerical simulation of crystal growth.
There is "Data" panel in LeoMonteCrystal to conveniently set thermodynamic and crystallographic parameters for numerical simulation of crystal growth.
The Fig. 1 shows screenshot of the LeoMonteCrystal “Data” panel.
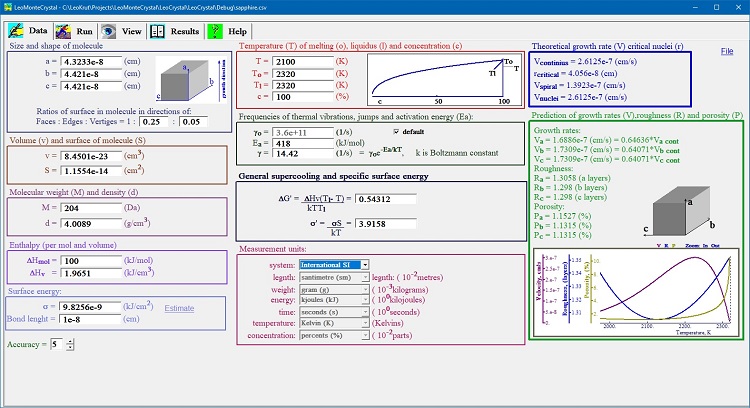
Fig. 1. Data panel contains groups of controls combined by functional
attributes for editing and displaying parameters of phase transition
these are directly necessary for setting numerical simulation
As a rule for changing any parameter user should to edit content of any parameter and press “Enter” button. All related to modified parameter variable will be changed automatically to preserve consistency of whole setting. It could be some irritations of unintentional value changes in attempt to use “Data” panel as invert calculator primary variables from higher order ones. As a rule if user inputs variable a entering staring from experimentally obtainable ones there should be no conflicts. To help user in keeping sign on all durative changes when editing control has focus all parameters these will be affected with upon change are background highlighted.
Size, shape and density of molecule:
|
|
||
| Fig. 2. Size and shape of molecule group. |
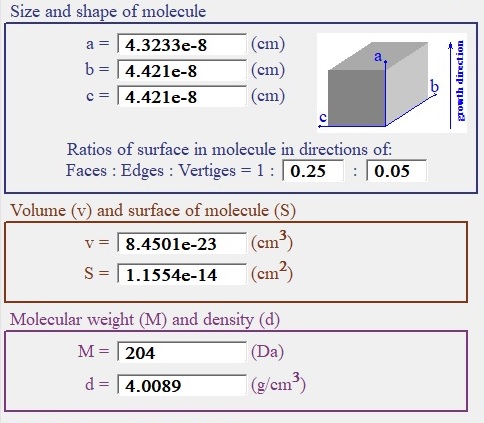
In LeoMonteCrystal parameters to most basic characteristics of molecules are corresponded to cell in the crystallographic lattice with orthorhombic group of symmetry. Size group shows also a shape of molecule for given sizes of cell cuboid and distribution of the surface of molecule between its faces, edges and vertices. The concept of uneven distribution of the surface along different directions are reflection anisotropy of molecular bonds in crystal. A representation of the molecule (structure element of the crystal lattice) as cuboid like structure element is made as best possible compromise between applicability of fast algorithm of cnumerical simulation and reasonable close representation of complexity and variety of structures of the crystals in reality.
Molecular weight here is measured in Daltons. (One Dalton (Da), approximately (but with more than enough preciseness for our goals) is 1/12th the weight of carbon-12 isotope.)
It is also important to distinguish between concepts of molecule (structure element) of crystal used here and unit cell of crystalline lattice in crystallography. There can be situation when one unit of lattice contains more than one molecule for example. The concept of the lattice unit is mostly related to exact atomic structure of the crystal. The idea of structure element (molecule) of the crystal is about most small element of the crystal that can be added to crystal with retaining all its properties. It is most commendable to be careful in selecting parameters of molecule for the simulation. Noncritical selection database crystallographic data for presentation of effective size and shape of molecule are with high probability can produce absurd results for crystal growth outcome during numerical simulation. Nevertheless there is direct connection between volume of cell in crystal lattice and volume of molecule here -their ratio has to be rational number.
Enthalpy and surface energy.
Fig. 3 shows controls permitting to edit thermodynamic parameters of crystallization. An enthalpy of crystallization is provided in two equivalent values: first is change of enthalpy of the system after one mol of crystal is appeared - ΔHmol (molar enthalpy); second that can be calculated from molecular enthalpy taking into account size of molecule and density of the crystal - ΔHv (volume enthalpy).
|
|
||
| Fig. 3. Enthalpy and surface energy group |

The formula:
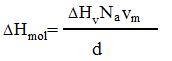
|
(1) |
represents connection between molar enthalpy - ΔHmol, and specific volume enthalpy ΔHv , where vm - a volume of one molecule, d - density and Na – Avogadro constant (6.0221×1023 mol-1).
There are two direct methods for independent measurements of molecular , ΔHmol, and specific, ΔHv, enthalpy. Specific enthalpy can be obtained by measuring latent heat during melting of the given amount of crystalline phase by methods of calorimetry that sometime is close to imposable to do. Molecular enthalpy can be calculated from phase diagram taking function of melting temperature of crystal versus concentration of crystal molecules in liquid phase it is in equilibrium with. Obtained independently together with experimentally measured density the direct information about volume and molecular weight of effective structure elements in crystal can be extracted to compare with crystallographic data obtained from X-ray diffraction.
Surface energy of the crystal contrary to the liquid does not manifest itself in almost any experiment except crystallization phenomena. The reverse analysis of numerical simulation of crystal growth finding value of σ that produce growth rate in agreement with experimentally observed one is one method to find value of surface energy. We offer the simple formula for primary estimation of the value of surface energy of crystal that is incorporated in LeoMonteCrystal. User can enter most probable effective length of chemical bonds between atoms on the molecule surface and click on "Estimate" link style button near surface energy to get result of our formula that taken into account enthalpy that must be set before.
Temperature, melting point, liquidus and concentration.
Fig. 4 shows group of controls represents experimental condition of growing crystal in non- stoichiometric melt.
|
|
||
| Fig. 4. Temperature, melting temperature, liquidus temperature and concentration group. |
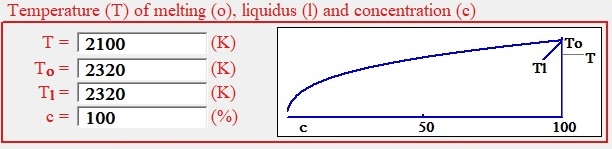
Let’s assume for specificity sake non-equilibrium crystallization of some salt in presence of other compounds needs to be researched. In general case pure salt has temperature melt is Ta. In solution with concentration of crystal molecules, c, the temperature of zero growth will be less than melting point and named temperature of liquidus, Tl. Growth of crystal will be at temperature T. The chart at Fig. 4 shows relations between temperature of liquidus and concentration of structure elements in solution for the given enthalpy of crystallization that is set as shown in corresponding control and calculated by the formula:.
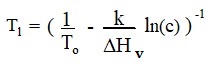
|
(2) |
Frequencies of thermal vibrations, activation energy and frequency of jumps.
Fig. 5 present kinetic characteristics of molecule jump in and out of crystalline phase in terms of model thermally activated reaction
|
|
||
| Fig. 5 Frequency of thermal vibrations, activation energy and frequency of jumps in crystal controls group. |
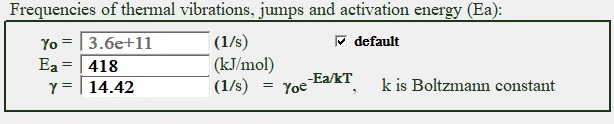
Parameters in this control group are not directly affecting a process of numerical simulation thus parameter γ serves as time scale coefficient for calculation of absolute value of growth rate. Activation energy usually in about ten times large (loose approximation) compare to enthalpy of crystallization and directly related to the energy of chemical bonding inside the crystal or liquid. If user choose to mark default box near the frequency control it will be estimated by the simple formula as velocity of the sound in solid (presumed as 1000 m/s) divided by size of molecule.
Specific supercooling and surface energy.
Fig.6 shows
|
|
||
| Fig. 6 Specific supercooling and surface energy controls group. |

a group of controls represented two generalized parameters with most directl influence on result of simulation. Specific supercooling ΔG` and specific surface energy σ` as can be seen from formula on the Fig. 6 are only two basic parameters defining probability for given molecule on surface to be emitted back into feeding growing crystal phase. Along size and surface distribution along of molecule these two variables unequivocally define result of simulation procedure.
Theoretical growth rate and size of critical nuclei.
The Fig. 7 shows theoretical growth rate
|
|
||
| Fig. 7 Theoretical growth rate and size of critical nuclei controls group. |

calculated for different most commonly discussed mechanisms for entered in Data panel parameters. Growth rate by continues growth mechanism Vcontinues is the maximum possible value for crystal growth rate and can be consider as measurement unit for all others mechanisms like two dimensional nucleation Vnuclei and spiral dislocation induced growth Vspiral. Formulas for calculation of these theoretical values can be found in our crystallization knowledge base.
Rates and roughness by state of art formulas and shape of nanocrystal.
Fig. 8 shows growth rates, roughness and porosity (concentration of the hole like defects) of during the growth of the crystal
|
|
||
| Fig. 8 Rates and roughness by state of art formulas and shape of crystal at the early stages. |
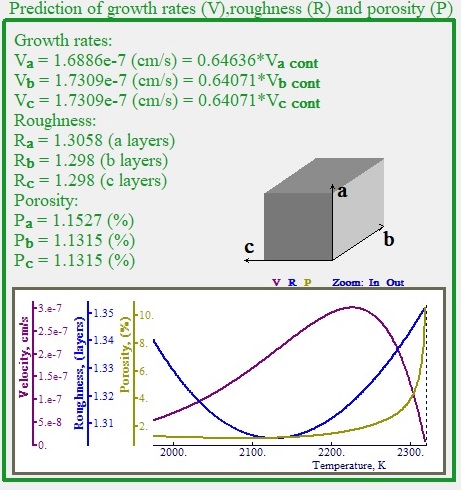
along all three major axes directions of the cuboid calculated from formulas derived by us from series of systematic numerical simulation of crystal growth made with help of LeoMonteCrystal. The shape of crystal on the very early stage of expansion (nano size) in case if surface energy of it is uniformly distributed along all direction is displayed as well. The temperature dependence of these parameters is shown on the chart.
Formulas for this calculations are quite complex, contains only mathematical constants as coefficients. These formulas was results of many years of devoted research and can be revealed for adequate compensation to interesting party as know-how transfer.
Measurement units.
Fig. 9 displays
|
|
||
| Fig. 9 Measurement units transferred controls group. |
“Measurements units” group of controls these permit to choose most custom for user selection of measurement units including non-standard ones. Thus “Data” panel can serve as stand alone unit converter software for resolving problems related phase transition phenomenon.
Saving and opening data set.
Fig. 10 shows
|
|
||
| Fig. 10 Saving and opening data set controls group. |
controls group to save in or open from comma delimited file initial parameters of "data" panel together with conditions of numerical simulation of "run" panel. The file can be edited in MS Excel that is although not recommended for not experienced user.
Accuracy.
Fig. 11 shows
|
|
||
| Fig. 11 Accuracy controls group. |
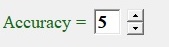
'accuracy" controls group permits to set number of meaningful digits presented in "data" panel. Four or five digits is normally sufficient or even redundant for most of practical cases but it's could be beneficial to be able to investigate some special cases in fine steps if there is could be very strong correlation from one of the parameters like surface energy.
Short key combinations.
In LeoMonteCrystal implements several short key combinations at keyboard for fast data manipulation:
Ctrl-s - will copy in clipboard short extraction of data (T - temperature, Vconc a, Vconc b, Vconc c theoretical growth in three axes directions in current set of size and time utits, Vpart a, Vpart b, Vpart c - scaling effect of surface energy on crystal growth rate in axis directions (unitless) and theoretical value of roughness of the surfaces perpendicular to growth axis - Sa, Sb, Sc in layers) to clipboard memory to be instantly paste in Excel.
Ctrl-t - will copy into clipboard memory title row for full report of the data including theoretical values.
Ctrl-c - will copy into clipboard memory row of full report of the data including theoretical values in current measurement units.
Ctrl-z - will calculate and put into clipboard memory spreadsheet like report when temperature of growth is varying from liquidus point down in current measurement units.
If active cursor is placed in one of the size of molecule controls combination Ctrl-number(1,2..9) or q will increase these molecule dimension scalable to this number or on 21/2 = 1.414214. Similarly Alt-number or q with scale down on these number. These operation will correct also molecular weight leaving density and specific volume enthalpy unchanged. It is convenient tool for fast research how size structure element (molecule) influences the mechanism, rate of crystallization in different directions and hence morphology of the crystals.