Physical background of the model for numerical simulation of crystal growth.
Physical background of the numerical model for crystal growth is described in details. There are definition of the simulation field and formulas for probabilities of elemental moves of molecules.
A model of growing crystal has to incorporate adequate mathematical presentation of the crystal and molecule moves applicable for software implementation. It has to reflect the molecule structure of the crystal surface including symmetrical positioning of molecules together with rules derived from atomic physics for calculation probabilities for each molecule to change their positions in small time step. Following is a description of the model used in software application LeoMonteCrystal.
Simulation field.
The physical reality of growing orthorhombic crystal is represented in the model by an integer matrix L[Nb, Nc] whose values are positions of the molecules on crystal surface measured in layers of crystallographic lattice in direction of the growth. Indexes of the matrix L give location of the projection of a molecule on the simulation field, where its dimensions, Nb and Nc, give a size of the simulation field. Fig.1 shows how for each element of matrix L these three numbers, one of them is for value of matrix and two others are for its indexes, unambiguously define a position of the molecule on the surface of the crystal. A value of the matrix element provides distance from base layer that also can be chosen as a starting position for the numerical experiment of growing surface. Two indexes represent projection of molecule on the base layer in the Cartesian coordinate system oriented consistently with orthorhombic lattice of the crystal. The positions of molecules in physical space can be obtained from integer values of matrix and its integer indexes, when all three of them are measured in layers of crystal, by scaling them by corresponding constants of crystallographic lattice.
|
|
|
|
|
|
Fig. 1. Visual presentation how two dimensional integer matrix represents a crystal surface in three dimensional space. Borders of active simulation field of matrix L are highlighted by blue color. |
|
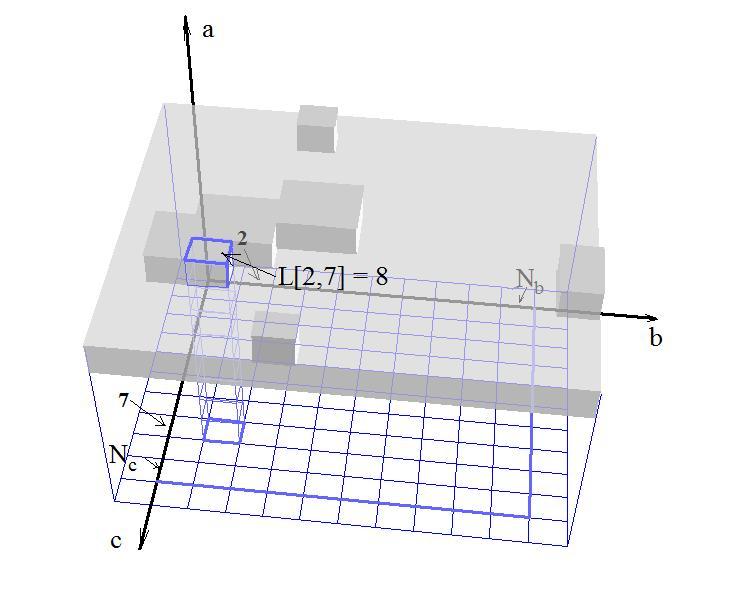
To avoid complications dealing with special conditions on edges of matrix L, elements at opposite sides of active field are glued to each other creating infinite kaleidoscope like space of simulation. It makes element L[i,Nc-1] neighbor to element L[i,0] and L[Nb-1,i] is touching L[0,j] (here and later we are using matrix L when zero based index notation is applied as it customs for C++ language that is used for software implementation).
In software implementation of the algorithm matrix L has two plus rows and columns: L[Nb+2, Nc+2] so next state of matrix L at first calculated on the active part of the matrix that doesn't include border rows and columns. Elements of these marginal rows and columns are taking into consideration nevertheless for calculating neighborhood of molecules inside of active field. After defining new values of elements in the active field these border elements are assigned to corresponding elements at active simulation area by following rules:
| for margin columns: | L[0,i]=L[Nb,i] and L[Nb+1,i]=L[1,i]; |
| for margin rows: | L[j,0]=L[j,Nc] and L[j,Nc+1]=L[j,1] |
In the Fig.1 borders of active simulation field of matrix L is highlighted by blue color.
Characteristics of the crystal surface these are in correspondence to the state of the matrix L can be calculated by the following formulas:
Position of the surface is defined as average value of all elements matrix L belonged to active field. It can be calculated by formula:
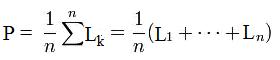
|
(1) |
where summing is performed for all n = Nb *Nc elementsof active field of the matrix L.
Average growth rate is defined by formula:
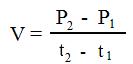
|
(2) |
where t1 and t2 are time checkpoints (arbitrary chosen) to calculate average rate of growth in between two time points during simulation experiment.
Roughness of the growing surface is calculated as standard deviation position of molecules from the average position P:
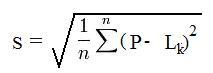
|
(3) |
If all elements in matrix L have the same value it means that the surface is 100% flat (absolute smooth). For absolutely flat surface the value of roughness equals zero (S =0.0). There is no theoretical limitation on the upper side of of the roughness parameter except applying general physical sense that suggests that more than ten layers roughness could be unrealistic.
Concentration of hole like defects below of the growing surface is calculated as ratio of number evens of creating hole Nh+ minus event to open existing hole Nh- to all position above base layer P*n.:
| Ch = (Nh+ - Nh-)/(P*n) | (4) |
The elementary event of creation hole like defect is formally associated with act of accepting a molecule at special bottle neck (well) like position in the matrix L. Such bottle-neck position in matrix Li is formally defined as having value by at least two less than all its closest neighbors. There is presumption that when molecule is incorporated in such position it does not move up to the bottom of "well" and leaves empty space below that (empty space) is interpreted as hole like defect. There is also an option of molecule above hole to be emitted from crystal open the hole is opened and a number of defects is lessened by one. During simulation every such elementary act is arithmetically summed to got actual number of hole defects below growing surface of the crystal.
An algorithm for numerical simulation is based on Monte Carlo implementation of modified cellular automaton model. The state of each element of matrix L for the next time step is changed randomly proportionally to calculated probabilities for corresponding molecule to make a jump into crystal or from it. These probabilities are calculated separately for each position at matrix L and is depending on relative positions of near neighbor molecules. For each molecule its nearest neighbors surrounding defines effective change of the surface of crystal in case of emitting molecule from crystalline phase into amorphous (noncrystalline) feeding matter surrounded crystal. A simulation algorithm can be described as infinite loop of calculations of next state of matrix L based on its current situation by set of rules these define probabilities for any given element of the matrix after certain period of time to change its position. There are three options for molecule at any give position after given time step, with the value set by researcher, to move ahead, back or stay at the same position. By application of random numbers generator one of these options is chosen proportionally corresponding probabilities for each element of the matrix to change position. That procedure will define state of matrix L at the next time step. Then this procedure repeats itself to calculate state of matrix L for next time step in infinite loop until reliable statistics of the process should be obtained.
Each element of matrix L represents corresponding position the surface of the crystal. At any given time step the value of element has an a choice to:
- Increase its value by one, that corresponds to the elementary act of incorporation into crystal a molecule from surrounding phase (melt, gas, plasma etc.) fed growth;
- Diminish its value by one that corresponds to the elementary act of the emitting of the molecule into the feeding surrounding;
- Retain its value.
- If researcher chooses to consider a possibility forming hole like defects during growth in a special situation when position of the molecule on surface is surrounding from all sides by walls of other molecules, forming well like construction. In such situation upon accepting a molecule that is sealing well atop a value the element of matrix L could move by two or more integer units ahead and also to form a hole type defect. Likewise when emitting a molecule opens existing hole style defect it is resulted in diminishing a value of element of matrix L by two or more integers.
First three options have probabilities p+, p- and po these are different for each element of matrix L. An actualization of the option number four (hole like defect) can be checked after accepting next molecule or emitting one. At any given time step in algorithm there are also two float (or in fact normalized to maximum random number integer) matrixes with the same size as matrix L for following probabilities:
- - first matrix, Po, contains probabilities for each given element not to move from current position at all, neither in or out;
- and
- - second matrix, P+, contains only probabilities for each element to move in.
Calculation of probabilities for move of each element.
In accordance to the theory of thermally activated reactions the frequency of incorporation of a molecule into crystal:
| γ+ = γo � e(-E a in/kT) | (5) |
and an opposite movement of molecule from the crystal into noncrystalline phase:
| γ- = γo � e(-E a out/kT) | (6) |
are defined by modified Arrhenius equation where γo - a frequency of thermal vibrations of a molecule, T - absolute (Kelvin scale) temperature, k - Boltzmann's constant (1.380 65×10-23 J K-1), Ea in and Ea out � energies for activation barriers for process of incorporation of the molecule into crystal and for opposite process of emitting it from crystal body into feeding phase correspondingly. The theory of thermally activated reactions postulates that molecule on surface to be incorporated into crystal must on the way to be deformed in some manner rearranging atoms into unstable intermediate atomic complex. Formation of such intermediate atomic complex (sometimes named activating complex) is accompanied by increase of free energy level during the jump (transformation) associated with the molecule. It's worth to note that jumps from crystalline phase and back not necessary associated with comparable to crystallographic lattice changing location of molecule in space on the scale of size of crystal layer. It can be accomplished by rearranging positions and bonds for neighboring atoms in location of molecule instead. The minimum necessary deformation is defined here is as such that demands a minimum of increasing energy level that permits to fulfill a phase transformation for single molecule.
A difference between energy levels of molecules in crystal and feeding phase is:
| Ea out - Ea in = ΔG�vm - σ�Δsm | (7) |
where ΔG is a change of specific Gibbs potential of free energy defined by equation:
| ΔG = ΔH* (ΔT / Tl) | (8) |
where ΔH � specific enthalpy of crystallization, ΔT
= Tl � T, where Tl � an equilibrium temperature of
the phase transition, T - temperature,
σ - surface energy of the crystal surface in contact with
surrounding phase, Δsm - change of surface of
the crystal after emitting of one molecule from any given position
on the crystal (note that for absolute rough surface in case
of continuous mechanism this value should be zero at average
and for smooth surface at layer by layer growth value of Δsm
should be significant large), vm � volume of molecule.
The implementation of the discussed model demands establishing a connection between frequencies of jumping of molecules in and out of crystal: γ+ , γ- with probabilities for molecule on surface in given position of matrix L to move ahead, back or stay on the same position: p+, p- and po.
Time step.
Average number of molecules that will be incorporated into surface of the crystal absent of the phenomena of emitting back, n+ , can be arbitrary defined by researcher as a starting point in defining value of time step. The value n+ should be small enough for adequate modeling of the process that should be achieved if random positions of these molecules n+ will fall far enough from each other not to be touching neighbors for every step. Other way around, there is the need for time step to be large enough to fit into always limiting computing resources. For specificity sake one can consider value about n+ = 10 on the simulation field with size n = 100*100 as quite proper compromise for both these demands for modern computers. The physical value of time step can be defined from the value of frequency of the processes of incorporation of molecule into crystal by formula:
| τ = (n+/n)/μ+ | (9) |
Probabilities for surface changing position on the molecular scale.
If value of time step approaches zero, τ →0, probabilities of given position on surface of the crystal to move on one step back or forth, or stay where it is can be calculated by equations:
| p+ = γ+ � τ = (n+/n) | (10) |
| p- = γ- � τ = p+�exp(-(ΔG�vm + σ�sm�Δsm)/kT) = p+�exp(-ΔG` - σ`�Δsm) | (11) |
| po = 1 - (p+ + p-) | (12) |
where general supercooling is defined as:
| ΔG` = ΔG�vm/kT | (13) |
and specific surface energy:
| σ` = σ�sm/kT | (14) |
The relative change of the crystal surface for event of emitting one molecule gives the formula:
| Δsm = (1 - 2 *scr) | (15) |
where scr is part of molecule on surface that is in direct contact with near neighbor molecules these are belonged to the crystalline phase.
Absent of limitations on computer's productivity and numerical precisions in calculations including random number generator formulas (10 - 12) can be used straightforwardly if small enough time step if chosen. Understanding that for Δsm → sm the value p-, calculated by formula 10, has largest possible value and arbitrary choosing p-(sm) ≡ 0.5 or lesser, we will be sure that any probabilities calculated by formulas (10 - 12) with
| n+ ≡ 0.5*n*exp(ΔG` - σ`) | (16) |
will be lesser than this arbitrary number (0.5). All calculated probabilities for movements of the molecules will be less than 1.0 and so will satisfy all the rules. There is large set of situations with proper combination of values of general supercooling and specific surface energy when such direct approach is perfectly workable. But not for every situation.
There are also infinite and most interesting set of big values of specific surface energy when this naive, direct approach, described above, cannot be realistically implemented even for modern computers. The problem manifests itself clearly in the fact that there always could be situations for large values of σ` when n+ should be too small for computers to handle. If value n for described direct approach had happened to be very small it produces at least too complications: first one is that time of numerical experiment will too consuming even most powerful computers to achieve practically applicable results. Second problem, that could even more problematic, occurs when precision of the generator of random numbers that can be defined as inverse value of its maximum produced number, 1/Nr_max, ought to be less or close to probability of one molecule jump into crystal per one time step. That condition can be presented by formula:
| n+/n ≥ 1/Nr_max | (17) |
We use two methods to expand area of workable ΔG`, σ` values beyond limitations presented by formulas 16 and 17. First one uses a model of quasi-independent behavior of molecule during one time step. The method permits to extend area of reliable results far beyond limitation of common sense condition that p- calculated by formula 11 must be always lesser than 1.0 for all possible situations. The model assumes that if one time step is divided onto infinite number of sub-steps, Nτ;, the result probabilities of one molecule to move can be independently defined assuming that all its neighbors would retain their positions during whole time step. The probability that the molecule will make at least one time jump from crystal into surrounding space can be calculated by the formula:
| p'- =1 - lim (1 - p-/Nτ)Nτ = 1 - exp(-p-) | (18) |
when number of sub-time steps approaches infinity, Nτ → ∞. Formula 18 produces obviously correct result as for very small and for very big values of p-.
The adequate design for the formula for probability of molecule to jump into crystal is not so obvious to obtain. We had to conduct numerical experiment for the quasi-static model when molecule starts from the position 0. It can jump:
- ahead (p+) to position 1 from position 0 or to position 0 from position -1;
- back (p-) to position -1 from position 0;
- back (p+-) to position 0 from position 1. It's important to note that probability p+- is analog of probability p- and calculated by the same formula 10 with the distinction that value Δsm corresponds to the situation when one molecule is moved ahead by one layer and all its neighbors stay at previous places. As a rule there is following inequality p+- ≥ p-.
One numerical experiment to research a model of quasi-independent behavior consists in repeating million (the large number the better) times a procedure whole for time interval, divided on the big number of equal sub-intervals with proportionally smaller probabilities (these must be less than 1.0) and register final position of molecule. The average positions at the end of whole interval after numerous tries represents probabilities to change position of molecule in one of described three manners. For given low value p+ between 0.0001 - 0.1 we found that formula (17) is very good ,almost perfect, representation of results of the numerical experiment for p'-. The probability of molecule to move one step forward is best described for such model by the fitting formula:
| p'+ = p+/(1 + Ko*(p- + p+-)K1) | (19) |
where numerical values of Ko = 0.6 and K1=1.33.
Formula 19 brings reasonable results for both borders for domain of values for p- and p+-, these can vary between 0 and ∞, that validates a structure of formula 19 in general. Nevertheless direct application of formula 19 in algorithm of crystal growth produces not satisfactory results for p+ with value more than 0.001 if coefficients Ko and K1 are set exact as they came from quasi-static experiment.
We found that the best way to incorporate formula 19 into algorithm is bringing an additional condition for defining coefficient K0 in formula 19 for given value K1. To define this special condition we postulate that the ratio of derived probabilities in the model of quasi-independent behavior of molecules to jump ahead and back should be the same as ratio of corresponding probabilities in formulas 9 and 10, when Δsm = sm (that is its larger possible value), and time step interval approaches zero:
| p'+/ p'-(sm) = p+ / p-(sm) | (20) |
Given that in case when Δsm = sm:
| p- (sm) = p+-(sm) = p+�exp(-ΔG` + σ`) | (21) |
and combining formulas 9, 10, and 18 coefficient Ko is defined by equation:
| Ko = 2-K1 p-(sm)1-K1/(1- exp(-p-(sm))) | (22) |
The best value for K1 in sense of maximum stability of results toward variation of p+ was found to be close to 1.1 Just as a wild hypothesis we set K1 = 1.1319882487943 that is a Divakar Viswanath's constant for growth rate of the random Fibonacci sequence. This hypotheses produces good results as well and we decided to stop on it.
Probability for molecule to finish at initial position is derived as rest of full set of variants:
| p'o = 1 - (p'+ + p'-) | (23) |
It's important to mention that probability of molecule not to do any moves at all is exp(-(p+ + p-)) that must be less than p'o as soon here is not taking into account moves back and forth. As a matter of abundance of cushions the condition that p'+ + p'- < 1.0 must be preserved and probabilities to be normalized if necessary.
The second method, used to expand a workable area for values of ΔG`, σ`, consists in performing for one set of initial parameters a series of numerical simulations runs varying only time step, or what is equivalent, probability for molecule to incorporate into crystal p+. The closer the value of p+ approaches zero the closer performance of the numerical simulation experiment will be to correct behavior leaving aside limitations of computer. The best way nearing to situation when p+ → 0 is an interpolation of produced results for series of different runs with different p+.
An interpolation method by itself can be sufficient in achieving adequate results without a model of quasi-independent behavior of molecules if it could be situation that function of any result parameter of the simulation like average growth rate or surface roughness would be linear or other easy fitted functions of p+. Unfortunately without applying special methods such functions can be significantly non-liner that emphasized necessity of usage combination of described above methods. We found that only in case of incorporation formulas from a model of quasi-independent behavior result functions of p+ can be easy fit for interpolation on p+ = 0 axis.
A part of molecule belonged to crystalline phase for given element of matrix L.
From previously presented formulas it is clear conclusion that parameter scr, part of the molecule surface in direct contact with its neighbor molecules these are belonged to crystalline phase, is a primary parameter for calculating probabilities of changing position of any given element of matrix L at next time step.
|
|
|
|
|
|
|
Fig. 2. Directions to near neighbors of molecule incorporated into a cell of orthorhombic lattice. |
|
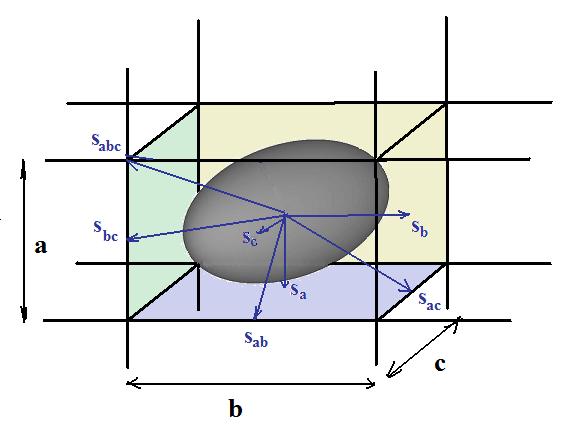
As it is shown in the Fig.2 a molecule inside of the cell of orthorhombic crystallography lattice has 26 unequally close neighbors along directions from its center toward its 6 faces, 8 vertices and 12 edges (6+ 8 + 12 =26). If surface of molecule are divided to areas associated with each neighbor molecules the value of molecule surface can be presented by equation:
| Sm = 2*(Sa + Sb +Sc) +4*(Sab + Sac + Scb) + 8 *Sabc | (24) |
where Sa, Sb, Sc � effective parts of molecule surface toward corresponding faces; Sab, Sac, Scb edges; and Sabc vertices.
There is other way to define surface of molecule as scaled surface of incorporated into cuboid cell molecule:
| Sm = 2* cs (a*b + a*c + b*c) | (25) |
where a, b, c � dimensions of the cuboid that is also crystallographic cell and cs � coefficient of proportionality of molecule's surface contained in cuboid. One can set cs = 1.0 presuming that surface of molecule coincides the surface of the cuboid that equivalent to the statement that effective surface energy of the molecule if its some smaller figure incorporated into cuboid must be scaled.
We are introducing following axiomatic in defining surface of the molecule. We define effective surface energy of molecule σ as such that should be if shape of molecule would be exact cuboid so cs = 1.0. We postulate also that edges and vertices of the presented by cuboid shape molecule have effective surfaces as defined in equation 24. The physical interpretation of the molecule surface associated with surface for formally one or zero dimension figures, as edges or vertices, is that atom bonds could have predominate their directions so breaking such bonds will release corresponding areas of molecule surface. We postulate that sf is parts of surface of cuboid like molecule associated with all its faces; se with all its edges; sv with all its vertices. Researcher can define them via to their relative values as shown in equation:
| sf : se : sv = 2*(Sa + Sb +Sc) : 4*(Sab + Sac + Scb) : 8 *Sabc | (26) |
Note that sum of values sf, se and sv equals 1.0:
| sf + se + sv = 1.0 | (27) |
Note that in specific implementation of our software LeoMonteCrystal user can define these parameters via two ratios toward of faces part of surface: Se= se/sf and Sv= sv/sf in form following ratios 1 : Se : Sv. In reverse taking into account equation 27 parts all relative parts of surface in these direction can be derived from these two ratios as following:
|
sf = 1/(1 +Se + Sv)
|
(28) |
It is logical to presume that relative input of different faces in whole molecule surface is proportional their individual surface:
| Sa : Sb : Sc = b*c : a*c : a*b | (29) |
In their turn we proclaim that relative surfaces associated with edges are proportional to their lengths:
| Sab : Sac : Scb = c : b : a | (30) |
All parts of surface associated with vertices have the same value.
Finally on the base of formulas 24 � 30 direct values of Sa, Sb, Sc, Sab, Sac, Scb, Sabc measured in unitless values as parts of whole surface of molecule - Sm can be calculated by following formulas:
|
Sa = (1/2) � cf � bc/(ab+bc+ac)
|
(31) |
Each element of matrix L representing molecule on surface of the crystal has 8 closest neighbor elements of matrix L. There is simple conditional algorithm that will be in details described in corresponding article that produces value of the scr, that is part of molecules surface in direct contact with crystal body, that is necessary to calculate probability for molecule to be out of molecule on the next step, p-, as combination of parts of molecule surface in different crystallographic directions defined in equations 31. The same part of surface of molecule that just be added on the tested position can be defined by this algorithm also to produce probability p+- that is needed in a model of quasi-independent behavior.
Summary. Matrix L permits adequate representation of the surface of orthorhombic crystal. On the base the theory of thermally activated reaction probabilities of the molecule to be incorporated and be emitted from crystal can be calculated for small enough time step for each molecule for any given position of molecule associated with element of matrix L taking into account values of the closest neighbors of the element to obtain values of molecule's surface in direct contact with crystal phase. A model of quasi-independent behavior of molecules during time step permits to expand areas of thermodynamic parameters in numerical simulation applicable for series of experiments run on modern computers for reasonable time frame.
Oct. 19, 2017; 12:46 EST